0:00 0:00
Article
NVIDIAがプロテオーム規模でタンパク質複合体を予測するGPUパイプラインを公開——AlphaFoldデータベース拡張へ
NVIDIAは2026年4月9日、Developer Blogでプロテオーム規模のタンパク質複合体構造予測パイプラインを公開しました。MMseqs2-GPUとNVIDIA TensorRT、cuEquivarianceを組み合わせ、AlphaFoldデータベースをホモ・ヘテロ複合体まで拡張する本格的なワークフローを示しています。
AlphaFoldデータベースを複合体まで広げる狙い
NVIDIAは2026年4月9日、NVIDIA Developer Blogにて「How to Accelerate Protein Structure Prediction at Proteome-Scale」と題した記事を公開しました。執筆はChristian Dallago氏らNVIDIAのバイオ・AIチームで、AlphaFold Protein Structure Databaseをホモマー・ヘテロマーの複合体まで拡張するためのGPU高速化パイプラインを解説しています。
記事ではまず、単体タンパク質の構造は公開データベースで網羅的に整ってきた一方、実際の生体機能を理解する上で不可欠な「複合体」の情報が依然として不足していると述べています。そこでAlphaFold-Multimerをベースに、STRINGから得られる相互作用データと組み合わせ、相互作用ペアを大規模に予測する仕組みを構築したとしています。
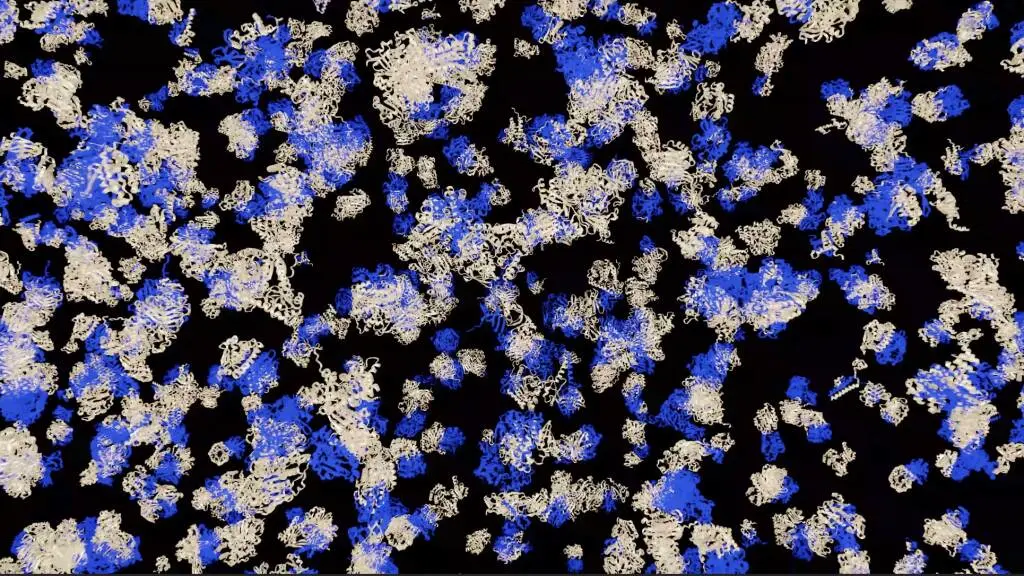
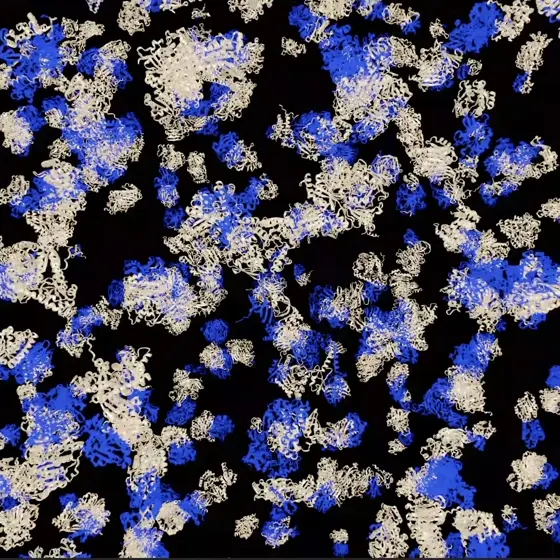
画像引用元: NVIDIA Developer Blog
MMseqs2-GPUとTensorRTによる高速化
パイプラインの中核は二段構成です。まずMSA(多重配列アラインメント)生成には、GPU最適化されたMMseqs2-GPUを採用しており、配列検索のボトルネックをGPU上でまとめて処理します。次に構造予測側では、NVIDIA TensorRTとcuEquivarianceによってディープラーニング推論をアクセラレートし、OpenFold実装上で同等精度を保ったまま大幅な高速化を実現したと紹介されています。
処理はNVIDIA DGX SuperPOD上のH100クラスタで、Slurmをジョブ管理に利用して走らせています。MSA生成と構造予測を別々のパイプラインに切り分けたことで、各段階を独立に最適化でき、計算リソースの効率的な使い分けが可能になったとNVIDIAは説明しています。
125ホモダイマーで73%が「利用可能」品質
ベンチマーク結果としては、125個のホモダイマータンパク質を対象にした比較で、ColabFoldと同等の予測精度を達成したと報告されています。具体的にはDockQスコア0.647を示し、73.0%が下流解析に利用可能な品質と判定されています。複合体予測では誤差が蓄積しやすい分、この水準を維持できる点はパイプラインとしての完成度の高さを示す指標と言えます。
スループット面では、ジョブの投入タイミングをずらす「staggered processing」によって最大25%の改善が得られ、1ジョブあたりのMSA配列数は300が最適値だったとされています。GPUを途切れなく稼働させるための細かな工夫の積み重ねが、プロテオーム規模で回すために効いているという構成です。
創薬・変異解釈・生成モデルへの応用
NVIDIAはこのパイプラインの応用先として、インターフェース部位に生じる遺伝子変異の解釈や、システム生物学的な構造解析、さらには創薬ターゲットの検証を挙げています。複合体インターフェースの情報は、単体構造だけでは見えない「どこに薬を効かせるべきか」の判断材料になるため、創薬の前段階における価値が大きいと紹介されています。
また、生成系のタンパク質設計モデルにとっても、高品質な複合体データはベンチマークや学習用データとして直接的な意味を持ちます。AlphaFoldデータベースを複合体まで拡張することで、RFdiffusionやChromaといった生成モデルの評価基盤を底上げできるとの示唆があり、NVIDIAは同パイプラインを「システム生物学から生成モデルまでをつなぐインフラ」として位置付けています。

How to Accelerate Protein Structure Prediction at Proteome-Scale
Proteins rarely function in isolation as individual monomers. Most biological processes are governed by proteins interacting with other proteins, forming protein complexes whose structures are…